Transcriptomics The Study Of RNA, Hussan Ahmad Ks [ebook reader browser .txt] 📗
- Author: Hussan Ahmad Ks
Book online «Transcriptomics The Study Of RNA, Hussan Ahmad Ks [ebook reader browser .txt] 📗». Author Hussan Ahmad Ks

TRANSCRIPTOMICS, PROTEOMICS,
STRUCTURAL AND FUNCTIONAL
PROTEOMICS, METHODS OF
STUDYING PROTEOMICS
TRANSCRIPTOMICS
Transcriptomics, the study of RNA in any of its forms.
The transcriptome is the complete set of transcripts in a cell and their quantity, for a specific developmental stage or physiological condition.
The transcriptome is the set of all RNA molecules, including mRNA, rRNA, tRNA, and other non-coding RNA produced in one or a population of cells.
TRANSCRIPTOMICS AIMS
To catalogue all species of transcripts, including mRNAs, noncoding RNAs and small RNAs.
To determine the transcriptional structure of genes, in terms of their start sites, 5′ and 3′ ends, splicing patterns and other post-transcriptional modifications.
To quantify the changing expression levels of each transcript during development and under different conditions.
TECHNOLOGIES
Hybridization-based approaches
fluorescently labelled cDNA with custom-made microarrays
commercial high-density oligo microarrays
Sequence-based approaches
Sanger sequencing of cDNA or EST libraries
serial analysis of gene expression (SAGE)
cap analysis of gene expression (CAGE)
massively parallel signature sequencing (MPSS)
APPLICATIONS IN PLANT BREEDING
Transcriptome assembly and profiling
The widespread use of transcriptome sampling strategies is a complementary approach to genome sequencing, and results in a large collection of expressed sequence tags (ESTs) for almost all the important plant species.
Small RNA characterization
Small RNAs (sRNA) are non-protein-coding small RNA molecules ranging from 20 to 30 nt that have a role in development, genome maintenance and plant responses to environmental stresses.
INTRODUCTION OF PROTEOMICS
Proteomics aims to identify all the proteins in a cell or organism including any post translationally modified forms, as well as their cellular localization, functions, and interactions
Understanding the proteome allows for:
Characterization of proteins
Understanding protein interactions
Identification of disease biomarkers
BIOMARKER
Biomarkers are biological indicators of a disease.
They are useful both for diagnosis, prognosis and response to therapy.
TYPES OF PROTEOMICS
Expression proteomics
Structural proteomics
Functional proteomics
Interaction proteomics
EXPRESSION PROTEOMICS
Expression proteomics is used to study the qualitative and quantitative expression of total proteins under two different conditions.
Normal and diseased state.
E.g : tumor or normal cell.
It studied that protein is over expressed or under expressed.
2-D electrophoresis.
STRUCTURAL PROTEOMICS
Structural proteomics helps to understand three dimensional shape and structural complexities of functional proteins.
It determine either by amino acid sequence in protein or from a gene this process is known as homology modeling.
It identify all the protein present in complex system or protein-protein interaction.
Mass spectroscopy is used for structure determination.
FUNCTIONAL PROTEOMICS
Functional proteomics explains understanding the protein functions as well as unrevealing molecular mechanisms within the cell that depend on the identification of the interacting protein partners.
So that detailed description of the cellular signaling pathways might greatly benefit from the elucidation of protein- protein interactions
INTERACTION PROTEOMICS
Proteins are not discrete and independent molecules they need other proteins or cofactors for their activity.
Such interactions are necessary for signal transduction, trafficking, cell cycle and gene regulation.
WHY PROTEOMICS?
Many types of information cannot be obtained from the study of genes alone. For example, proteins, not genes, are responsible for the phenotypes of cells. It is impossible to elucidate mechanisms of disease, aging, and effects of the environment solely by studying the genome.
TECHNIQUES IN PROTEOMIC STUDY
2-D electrophoresis
MALDI
Mass spectrometry
PMF(peptide mass fingerprinting)
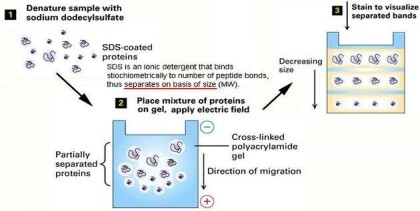
2-D ELECTROPHORESIS
Electrophoresis is the migration of charged molecules, particles or ion in a liquid or solid medium under the influence of an electric field.
1969 - introduction of denaturing agents especially SDS separation of protein subunit ( Weber and Osborn ).
This technique combines the technique IEF (first dimension), which separates proteins in a mixture according to charge, with the size separation technique of SDS-PAGE (second dimension).
The combination of these two technique to give 2-D PAGE provides a highly sophisticated analytical method for analyzing protein mixtures.
2-DE
Several forms of PAGE exist and can provide different types of information about the protein(s).
SDS-PAGE (sodium dodecyl sulphate-polyacrylamide gel electrophoresis), the most widely used electrophoresis technique, separates proteins primarily by mass.
Non denaturing PAGE, also called native PAGE, separates proteins according to their mass: charge ratio.
Two-dimensional PAGE (2D-PAGE) separates proteins by isoelectric point in the first dimension and by mass in the second dimension.
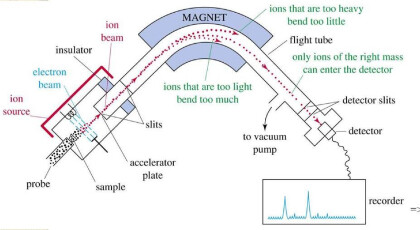
MASS SPECTROMETRY
MS/MS plays important role in protein identification (fast and sensitive).
Derivation of peptide sequence an important task in proteomics.
Derivation without help from a protein database, especially important in identification of unknown protein.
MAJOR PARTS
Sours ionized the sample
Analyzer separate the ions on m/z ratio
Detector sees the ions and analyzed the result
MASS SPECTROMETER
STEPS
Isolate cell or other protein source
Lyses cells and isolate proteins
Break up proteins into smaller (but still relatively large) amino acid chains
Separate chains (2D gel, gas or liquid chromatography)
Analyze separated protein parts by mass spectrometry
MASS SPECTRUM
Proteins consist of 20 different types of amino acids with different masses (except for one pair Leu and Ile).
Different peptides produce different spectra.
Use the spectrum of a peptide to determine its sequence.
PEPTIDE MASS FINGERPRINTING
Protein is cleave in smaller peptides.
Masses measured with MS.
These masses are then compared to known protein.
Computer programs translate the known genome of the organism into proteins.
Cut the proteins into peptides and calculate the masses of peptides.
Compare the known and unknown protein.
MATRIX ASSISTED LASER DESORPTION/IONIZATION(MALDI)
By pulses of laser light on the sample, ions of interest formed.
Large bio molecules can be determined and synthetic polymer greater than 200,000 Dalton.
Advantage
High speed
Relative immunity to contaminants
TIME OF FLIGHT
MALD with MS is called time of flight or TOF. This enables accurate and fast molar masses determination along with determined impurities and sequencing repeated units.
It is a method of mass spectrometry in which an ion’s mass-to-charge ratio is determined via a time measurement
ISOTOPE-CODED AFFINITY TAG(ICAT)
Label protein samples with heavy and light reagent .
Reagent contains affinity tag and heavy or light isotope.
Chemically reactive group: forms a covalent bond to the protein or peptide.
Isotope-labeled linker: heavy or light, depending on which isotope is used.
Affinity tag: enables the protein or peptide bearing an ICAT to be isolated by affinity chromatography in a single step.
PRINCIPLES
The peptide sequence of the protein to be quantified (between 5-25 Amino acids long)
contains sufficient information
Peptides tagged with the light and heavy reagents respectively are chemically identical.
STEPS OF ICAT
The principles of Isotope coded affinity tags are divided into four stages.
Sampling
Tagging
Isolation
Quantification.
ICAT is an in vitro labeling procedure that involves tagging of protein or peptide samples with the ICAT reagent specifically at their Cys residues. The ICAT reagent consists of a biotin tag, a light or heavy linker chain and a Cys-reactive group. One sample is tagged with the light ICAT reagent while the other is tagged with heavy ICAT.
SAMPLING AND TAGGING
Protein samples
Side chains having reduced cystein residues
Through breaking cell structure
Samples are tagged with light isotope and heavy isotope
Light form of the ICAT reagent (containing zero deuterium)
Heavy form of ICAT reagent (containing eight deuterium)
Combine both into one complex mixture
Protease act to break
Larger protein molecules convert into smaller peptide
PEPTIDE ISOLATION
Avidin is then introduced to isolate the ICAT-tagged peptides from the mixture through affinity chromatography.
The isolated peptides are then analysed and separated by micro-capillary high performance liquid chromatography- mass spectrometry (HPLCMS/MS).
PROTEIN QUANTIFICATION
Quantification is achieved by comparing the integrated peak intensities for simultaneously eluted pairs of identical, doubly charged peptide ions.
The chemically identical ICAT-labelled peptide ions are readily identified because as they co-elute, they differ in mass-to-charge (m/z) ratio because of an 8 deuterium difference in the mass of the ICAT reagents.



Publication Date: 12-29-2020
All Rights Reserved
Dedication:
Book By Hussan Ahmad Ks •Transcriptomics, the study of RNA in any of its forms. •The transcriptome is the complete set of transcripts in a cell and their quantity, for a specific developmental stage or physiological condition. •The transcriptome is the set of all RNA molecules, including mRNA, rRNA, tRNA, and other non-coding RNA produced in one or a population of cells. TRANSCRIPTOMICS AIMS •To catalogue all species of transcripts, including mRNAs, noncoding RNAs and small RNAs. •To determine the transcriptional structure of genes, in terms of their start sites, 5′ and 3′ ends, splicing patterns and other post-transcriptional modifications. •To quantify the changing expression levels of each transcript during development and under different conditions.





Comments (0)